收稿日期:2020-12-21
基金项目:上海市青年科技英才扬帆计划资助项目(21YF140800)
作者简介:胡旭(1990—),男,讲师,博士,研究领域为发动机内燃烧与流动仿真。
基金项目:上海市青年科技英才扬帆计划资助项目(21YF140800)
作者简介:胡旭(1990—),男,讲师,博士,研究领域为发动机内燃烧与流动仿真。
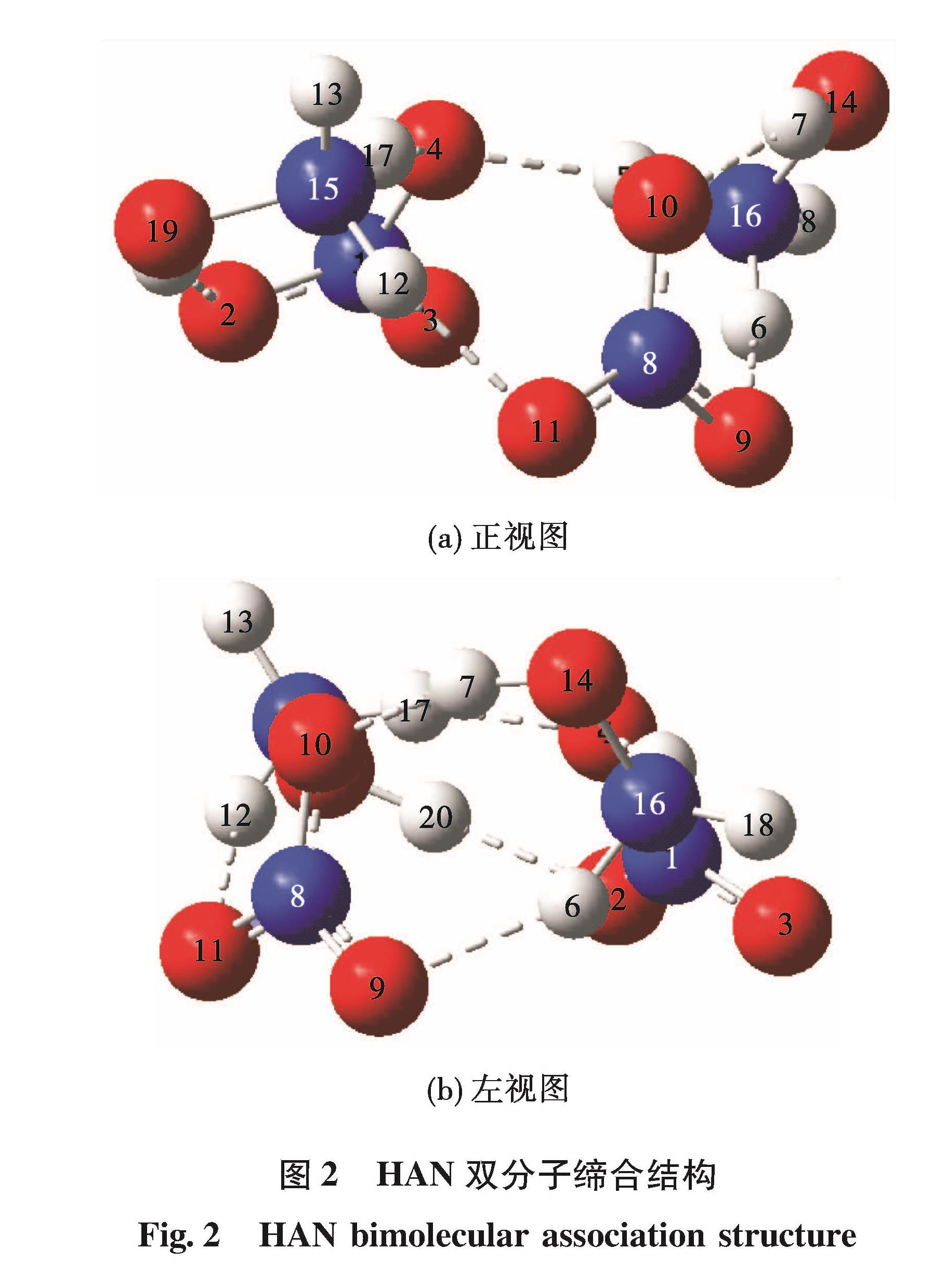
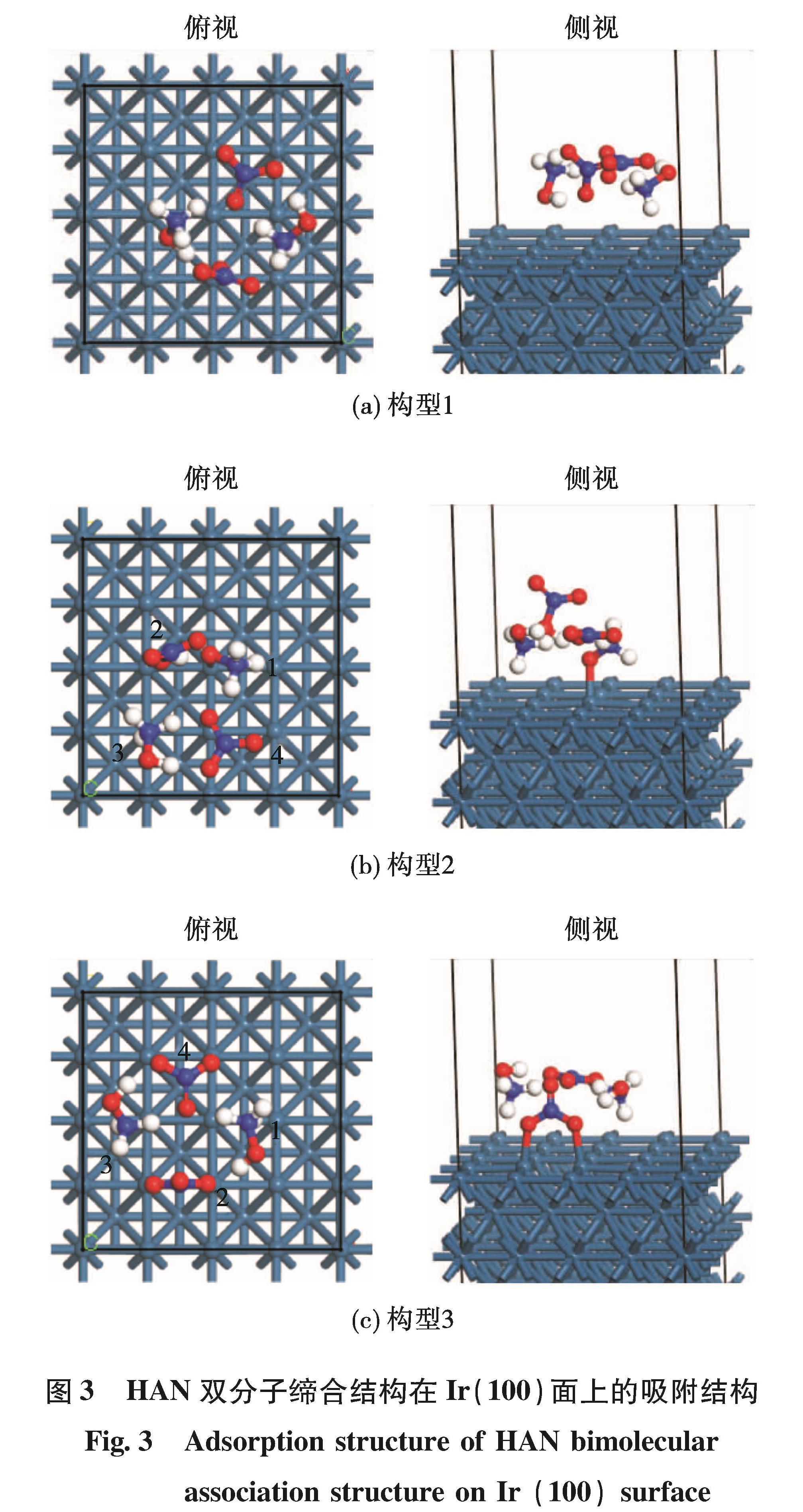
为揭示单组元液体火箭发动机内硝酸羟胺(HAN)基推进剂的催化分解反应过程,采用从头算算法确定HAN及其缔合物结构,利用密度泛函方法对稳定的HAN双分子缔合结构在Ir(100)面上的初始催化分解反应机理开展研究。计算结果表明:HAN分子是由双点位氢键结合而成的,其缔合结构主要是依靠分子间的氢键作用而成的。HAN双分子缔合结构在Ir(100)面上共存在有3种吸附结构,其中通过O与Ir键合作用形成吸附结构较为稳定,吸附能分别为-1.64 eV和-2.15 eV,而氢键作用结合的吸附结构的吸附能仅为-1.12 eV。HAN双分子缔合结构在表面发生的分解反应为放热反应,双分子缔合结构中2个HAN分子的分解反应发生存在先后顺序,不同的吸附构型对应不同的分解产物。在羟胺吸附结构中,其分解产物为HAN、NO-3、OH和NH3,而在硝酸根吸附结构中,其分解产物为HAN、NH3OH+、O和NO2。两种吸附构型下的分解能垒分别为12.68 kcal/mol和11.30 kcal/mol,在催化分解反应过程中这两种分解反应会同时发生。
The catalytic decomposition mechanism of hydroxylamine nitrate(HAN)bimolecular association structure on Ir(100)surface was studied to reveal the catalytic decomposition processing of HAN-based propellant in momopropellant liquid rocket motor.The Ab Initio method was adopted to obtain the molecular structure of HAN and its bimolecular association.The catalytic decomposition process was calculated by density functional theory method.The calculation results indicate that the HAN molecule is formed by double point hydrogen bonding and the association structure is mainly formed by hydrogen bonds between molecules.Three adsorption structures of HAN bimolecular association on Ir(100)surface was found.The adsorption structure is stable under the cooperation of O and Ir bonds,and the adsorption energies are -1.64 eV and -2.15 eV,respectively.The adsorption energy under the hydrogen bonded adsorption structure is only -1.12 eV.The decomposition reactions of HAN bimolecular association structure on the surface are all exothermic.The decomposition of two HAN molecules in bimolecular association happens under different order.With different adsorption structure,the products of the decomposition are different.In the hydroxylamine adsorption structure,the decomposition products are HAN,NO-3,OH and NH3,while in the nitrate adsorption structure,the decomposition products are HAN,NH3OH+,O and NO2.The decomposition energy barriers of the two adsorption structures are 12.68 kcal/mol and 11.30 kcal/mol,respectively.These two reactions are all available during the catalytic decomposition processing.