2.1 JP-10燃烧过程反应能量变化特征
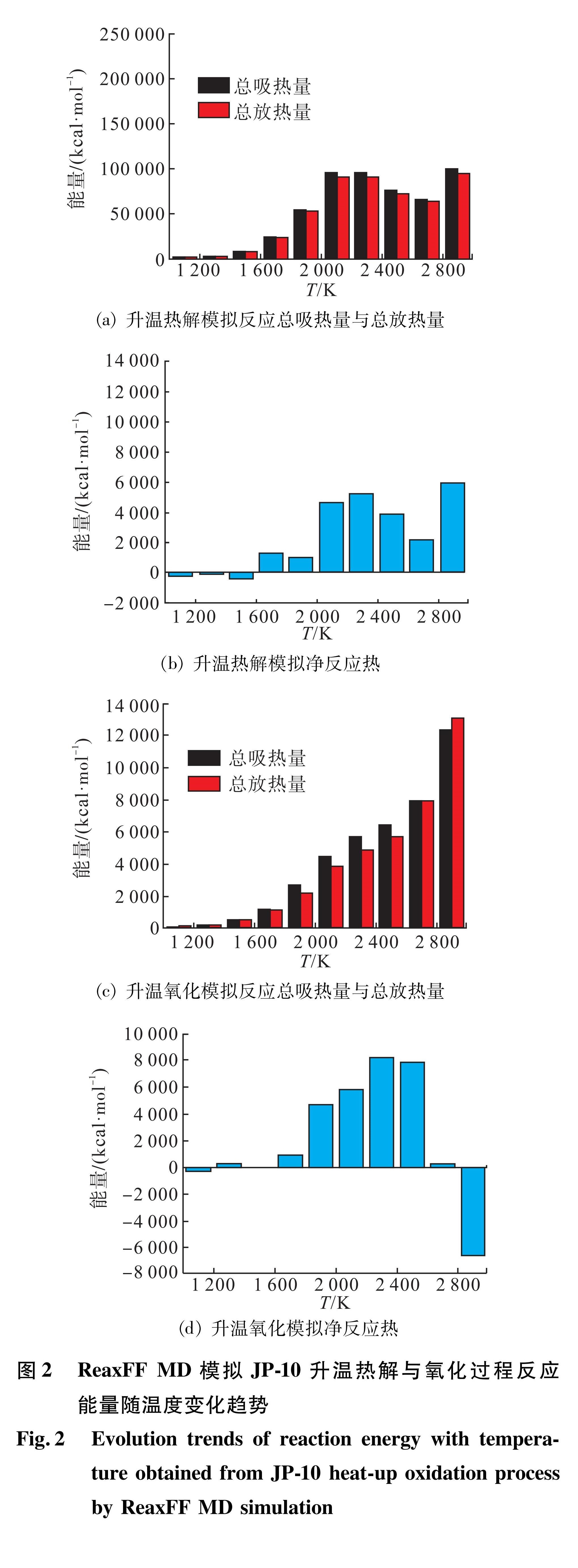
碳氢燃料高温反应中微观吸热和放热反应是耦合伴随的复杂过程,为了分析燃料主要吸热反应的类型与发生的阶段,通过1 000~3 000 K之间JP-10分子体系的升温热解和升温氧化模拟分别获得了体系总反应能量随温度的变化趋势。图2以温度区间为单元划分为10段,以此考察了每个温度区间内的反应热。图2(a)与图2(c)结果显示,燃料燃烧过程在微观上的吸热和放热反应的总吸热量和总放热量的数值远高于净反应热,这是由于振荡反应造成的,即微观的单个反应会在正向和逆向反应过程中重复多次才能生成最终产物。由图2(b)与图2(d)可知,热解体系中总反应热效应体现为吸热,而氧化体系中总反应热效应体现为先吸热后放热。两者的区别主要体现在2 600 K之后,氧化体系中由于热解产物被逐渐氧化,造成总反应吸热量降低并转变为放热。
图2 ReaxFF MD模拟JP-10升温热解与氧化过程反应能量随温度变化趋势
Fig.2 Evolution trends of reaction energy with temperature obtained from JP-10 heat-up oxidation process by ReaxFF MD simulation
模拟结果表明:JP-10升温氧化产生的自由基种类繁多,主要有H、HO、HO2、O、CH3、C2H3、C2H3O、C2H3O2、C2H5、C2O2、C2HO、C2HO2、C3H3、C3H3O、C3H3O2、C3H5、C3H5O、C3H5O2、C4H5、C4H5O、C4H7、C4H7O、C5H5、C5H5O、C5H7、C5H7O、C6H7、C6H9、C7H11、C8H11。其中数量较多的自由基为HO、HO2、C2H3、C3H5、C5H7,且含氧自由基均为C5及以下分子。稳定气体产物分子主要有C2H2、C2H4、C3H4、C4H6。因此本文认为,JP-10燃料发生开环反应后,C10分子裂解至C5分子过程基本不发生氧化反应; 氧化反应主要发生在C5及以下的小分子产物中,且C1-C3分子的氧化反应占主导地位。考虑到JP-10升温氧化过程中C10分子裂解至C5分子过程基本不发生氧化反应,与JP-10纯热裂解模拟得到的结果相一致[20],表明JP-10燃料的化学吸热来源于初始开环、裂解、深度裂解和脱氢反应,实际吸热量取决于燃料裂解的程度。JP-10分子氧化过程从吸热到放热的转折源于C5以下小分子的深度裂解反应与氧化反应的竞争。基于JP-10热裂解吸热与氧化放热历程的特性,可以将JP-10氧化过程中的吸热与放热耦合现象进行解耦研究,为此采用慢速升温热解模拟(1 000~3 000 K,2 K/ps)分析JP-10的热解吸热反应机理,采用长时间恒温模拟(3 000 K,2 ns)分析JP-10的氧化放热反应机理。
2.2 JP-10热解吸热反应机理
通过JP-10升温热解模拟得到的6类主要反应分别为JP-10的初始开环反应、中间产物的后续开环反应、裂解反应、脱氢反应、氢转移反应和加氢反应。其中加氢反应的整体热效应为放热,剩余5类均为净吸热反应,具体的主要反应类型、平均反应热、反应数量和计算得到的总反应热如表1~表3所示。本文为了使反应式表达式简练,末端和自由基位点的H显式标出,其他位置的H未标出。
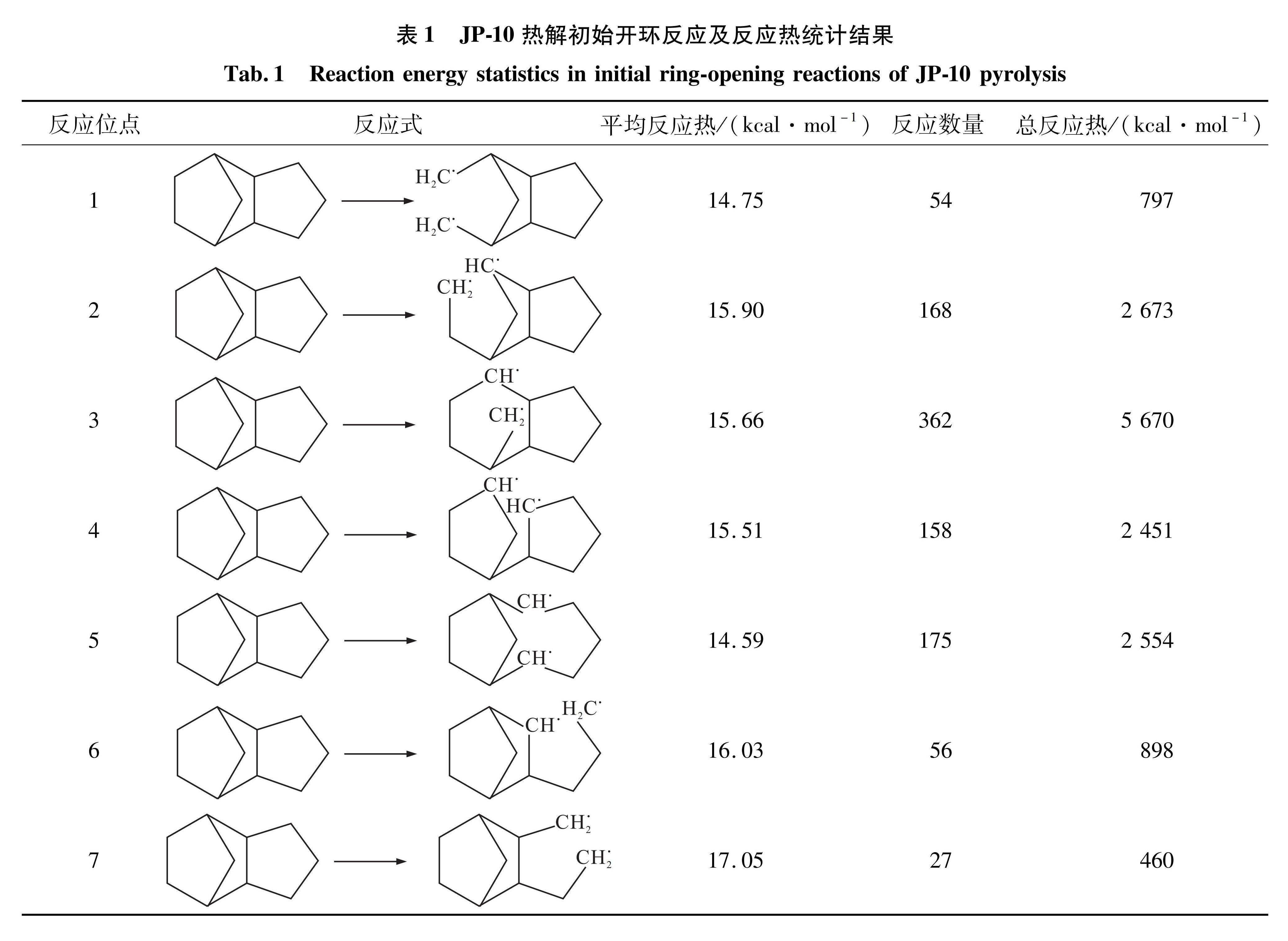
表1 JP-10热解初始开环反应及反应热统计结果
Tab.1 Reaction energy statistics in initial ring-opening reactions of JP-10 pyrolysis
由于JP-10的第一步开环反应缺乏活性自由基的诱导,需要吸收大量的热量才能使C-C单键断裂即发生单分子裂解反应。表1列出了JP-10分子7个位点的开环反应的平均反应热和反应数量,总吸热量为15 503 kcal/mol。结合图2(b)中结果提示的JP-10在2 000~2 600 K之间热裂解反应的吸热量为14 038 kcal/mol,主要由初始开环反应所贡献。
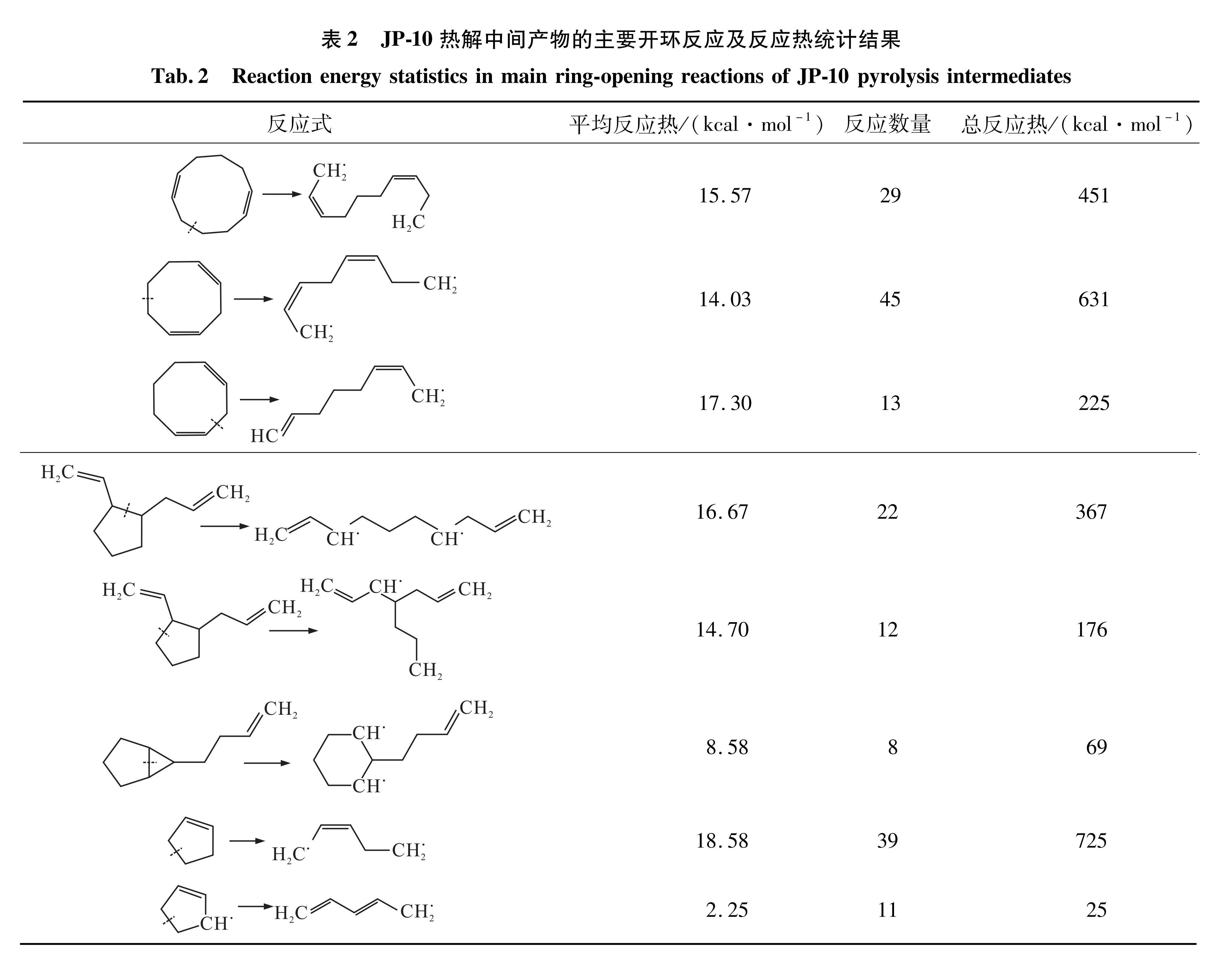
表2 JP-10热解中间产物的主要开环反应及反应热统计结果
Tab.2 Reaction energy statistics in main ring-opening reactions of JP-10 pyrolysis intermediates
JP-10热解中间产物主要有1,6-环葵二烯、1,4-环辛二烯、环戊烯和多种带支链环戊烷,进一步开环反应生成C5-C10直链和支链烃,如表2所示,总热效应为2 669 kcal/mol。由于这些环烃还可能发生其他类型的开环反应,且具有较多的同分异构体,除了直接开环还可能先断裂支链,因此模拟中的后续开环反应吸热量更高。
表3 JP-10热解模拟中的主要裂解、脱氢和氢转移反应及反应热统计结果
Tab.3 Reaction energy statistics in main pyrolysis, dehydrogenation and hydrogen transfer reactions of JP-10 pyrolysis
如表3所示,JP-10开环产物的裂解反应趋势是C8→C5+C3、C5→C3+C2、C4→C2+C2、C3→C2+C1,主要发生在2 600 K之后。表3中4类裂解反应的总热效应为3 164 kcal/mol。C8链烃以辛三烯为主,由JP-10分子经过多步开环、脱乙烯所生成; C6和C7链烃数量较少,因此未列出; C5链烃主要是戊二烯,由C8链烃裂解和环戊烯开环生成的C5H7(戊二烯自由基)加氢所生成,C5H7会裂解生成C2H2和C3H5自由基; C4物种主要是丁二烯,由JP-10恒温热解机理结果[20]可知,丁二烯在高温条件下会裂解为乙烯基; C3物种主要是C3H5(丙烯基),有3种同分异构体,容易发生裂解反应的结构是CHCHCH3,其继续裂解会生成乙炔和CH3自由基,该反应是CH3的主要来源。表3中3类主要脱氢反应总热效应为6 104 kcal/mol,是2 600 K后高温条件下最主要的吸热反应。碳氢小分子自由基能够通过脱氢反应生成稳定Π键与H自由基,JP-10热解体系的主要自由基为CH3、C2H3、C3H5和C5H7,其中CH3较难脱氢生成亚甲基CH2、C5H7更易发生裂解反应,因此脱氢反应主要发生在C2H3和C3H5自由基上。表3中2类主要氢转移反应热效应为990 kcal/mol。例如,C5H7和C3H5的氢转移反应均为伯碳自由基转变为更稳定的仲碳自由基的过程,因产物在高温条件处于能量较高的构象,使得反应热效应体现为吸热,不同于常规认识。
表1~表3的数据表明:JP-10的开环、裂解、脱氢与氢转移反应的总吸热量为28 430 kcal/mol; JP-10的初始开环反应与后续开环反应的总吸热量为18 172 kcal/mol,约占总吸热量的64%,且JP-10在2 600 K之前发生的吸热反应主要为初始开环反应。
2.3 JP-10氧化放热反应机理
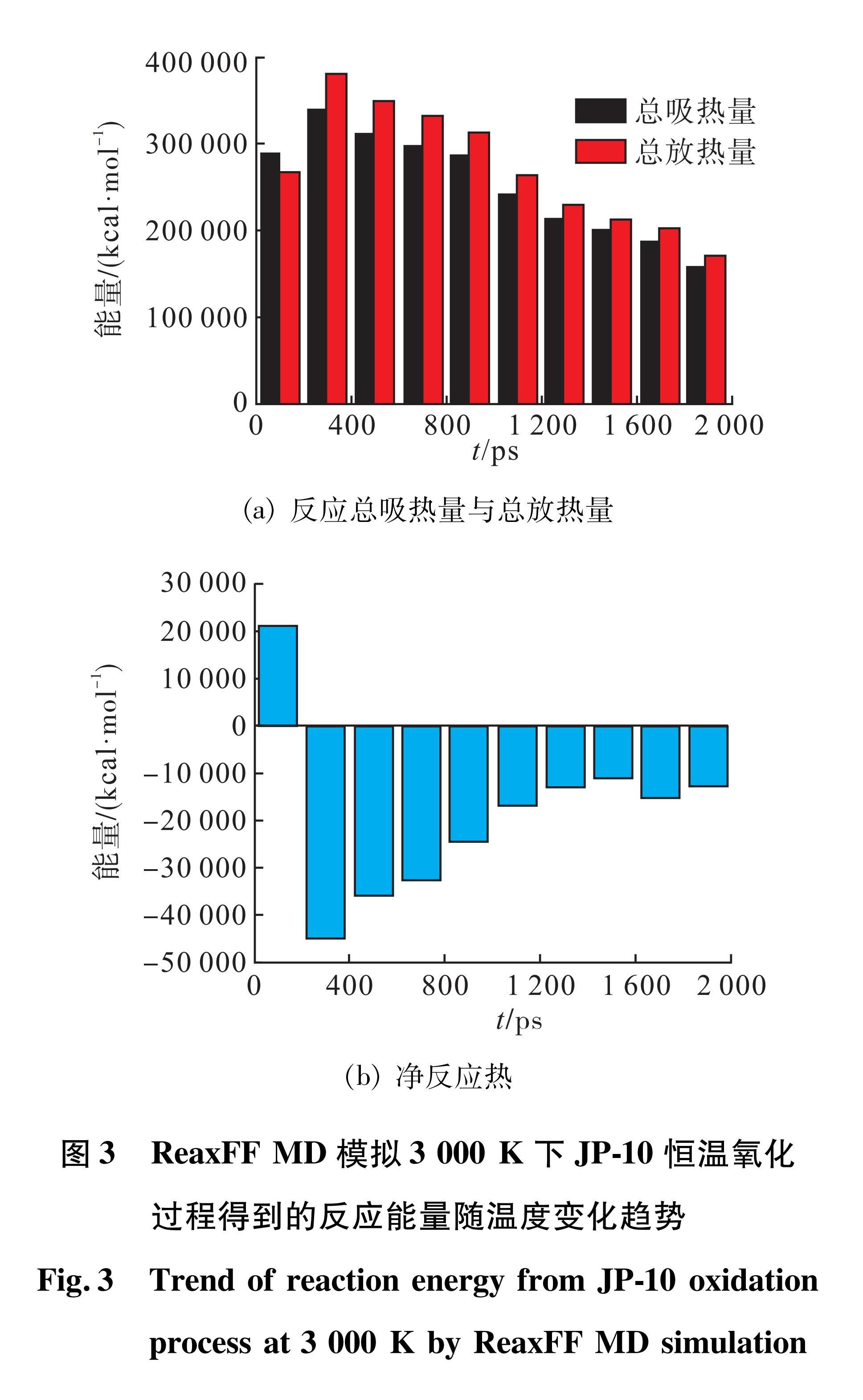
由于碳氢燃料的氧化反应的发生相对于热解反应需要更长时间及更高温度,因此利用3 000 K、2 ns模拟时长和当量比为1的恒温模拟分析JP-10的氧化反应机理,该模拟条件下O2消耗量达到82.7%,相应的CO2产率为73.9%,H2O产率为82.3%,可以认为氧化反应进行得相当充分,方便考察比较完整的氧化反应历程。模拟结果显示,H2O的生成时间比CO2更早,而H2数量很少。JP-10氧化过程中会生成大量的氧化中间产物,数量最多的为CH2O(甲醛)、HCCOH(乙炔醇)、H2CCO(乙烯醛)、HOCCOH(乙炔二醇)、OCCO(乙烯二醛)。HO与HO2是氧化过程中最重要的两种自由基,最高可占自由基总量的45.14%,在500 ps左右达到峰值。JP-10分子体系总反应能量随温度的变化趋势如图3所示,反应前200 ps内由于裂解吸热导致净反应热为正,200 ps后开始大量放热,放热量随时间减少,与HO和HO2自由基的演变趋势一致。
图3 ReaxFF MD模拟3 000 K下JP-10恒温氧化过程得到的反应能量随温度变化趋势
Fig.3 Trend of reaction energy from JP-10 oxidation process at 3 000 K by ReaxFF MD simulation
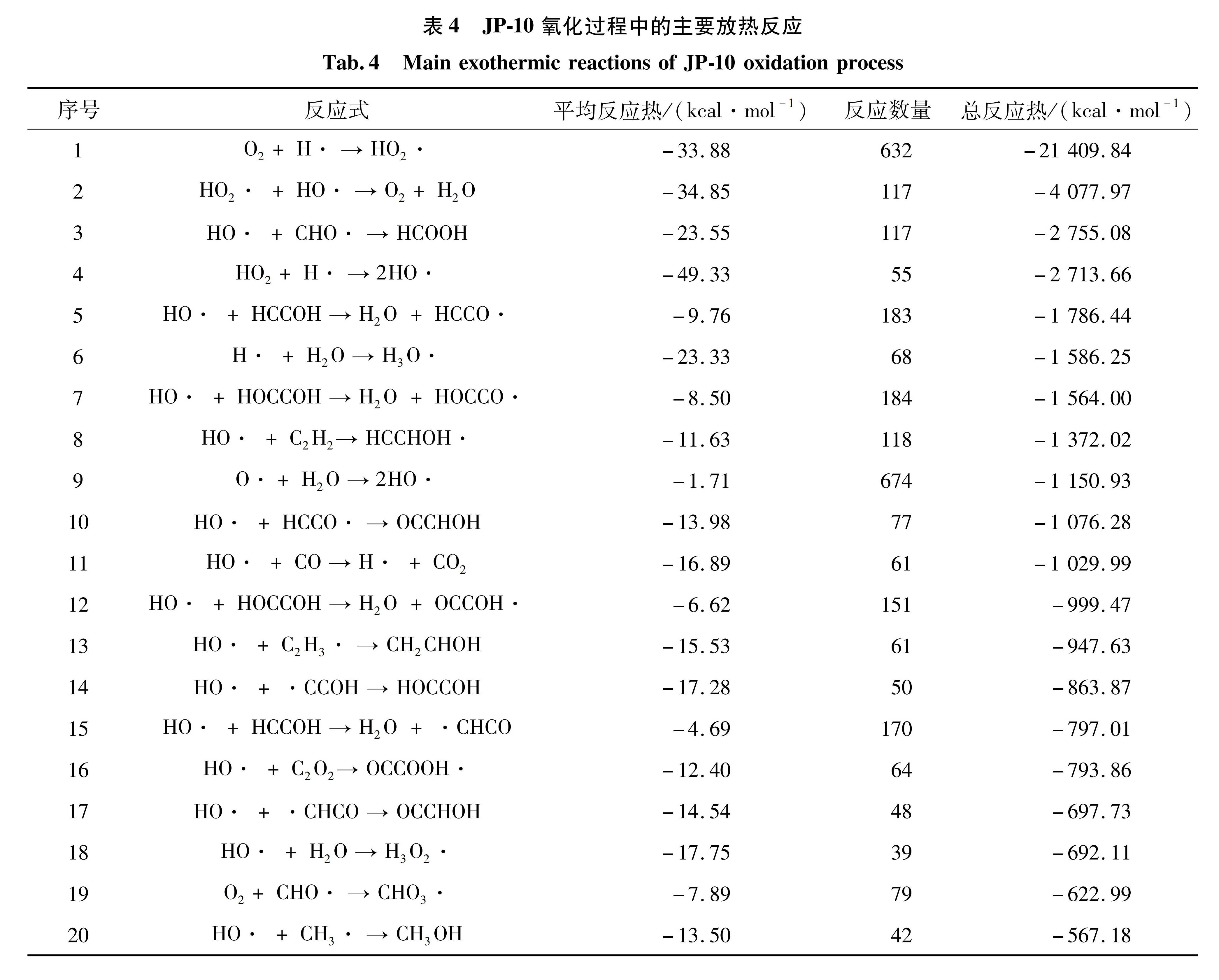
JP-10恒温氧化模拟中放热量最高的20个反应和对应数量、反应热如表4所示,总放热量为-47 504.31 kcal/mol,其中H和O2生成HO、HO2自由基和H2O的反应的总放热量为-25 487.81 kcal/mol,约占总放热量的54%。
表4 JP-10氧化过程中的主要放热反应
Tab.4 Main exothermic reactions of JP-10 oxidation process
JP-10的氧化反应主要发生在C0-C3物种之间,按照反应历程的顺序可以概括为如下步骤。
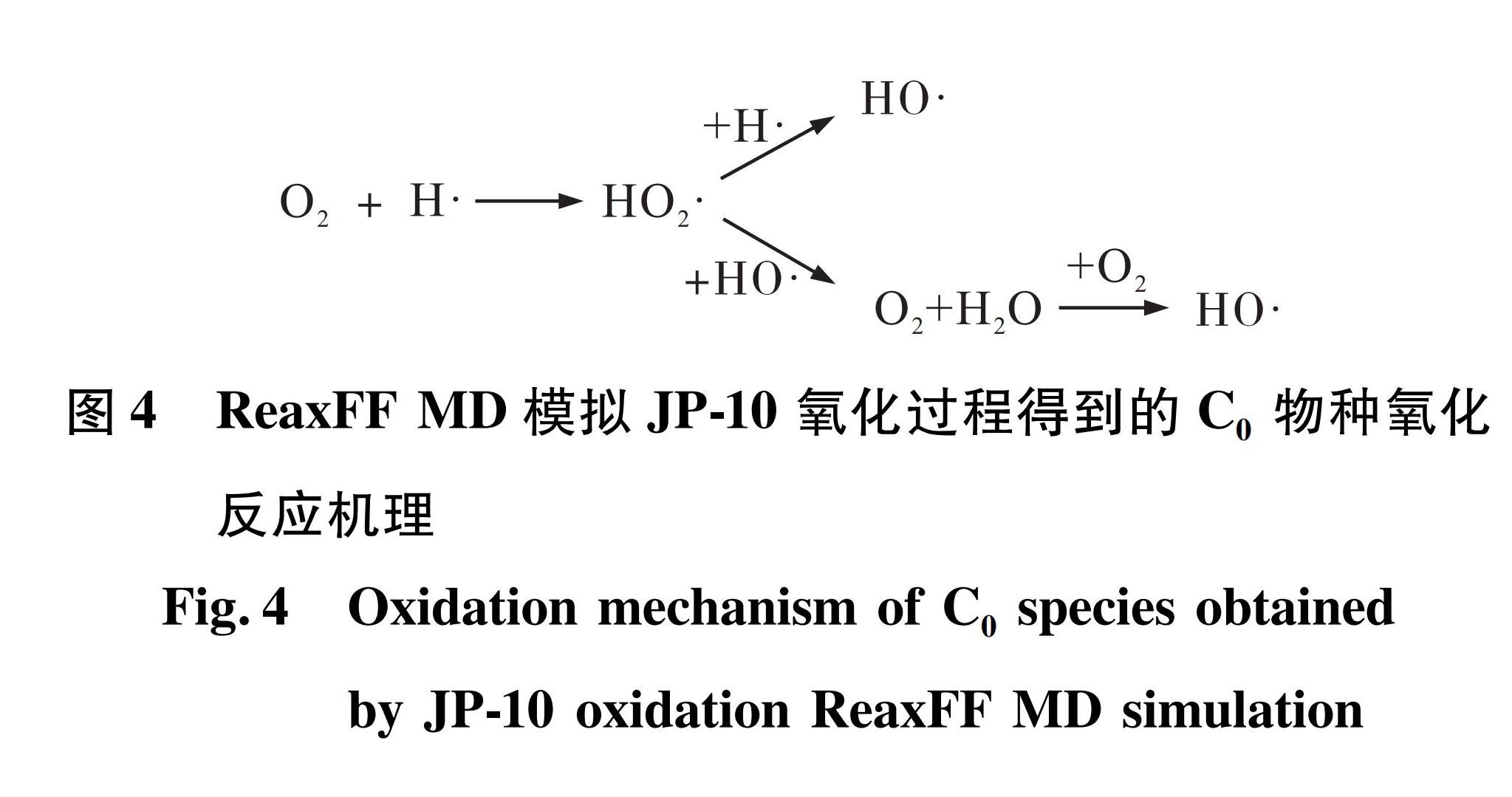
1)体系中的各类分子发生脱氢反应后产生H自由基作为引发剂。H可以由JP-10热解中间产物和氧化中间产物脱氢生成。O2和H发生多步反应后先生成HO2,最终生成HO,如图4所示。当HO浓度较高时,HO2可以与HO生成终产物H2O,这是H2O生成较早的原因。在高温高压的模拟条件下,H2O也可能与O2反应生成HO,作为新的链引发反应。
图4 ReaxFF MD模拟JP-10氧化过程得到的C0物种氧化反应机理
Fig.4 Oxidation mechanism of C0 species obtained by JP-10 oxidation ReaxFF MD simulation
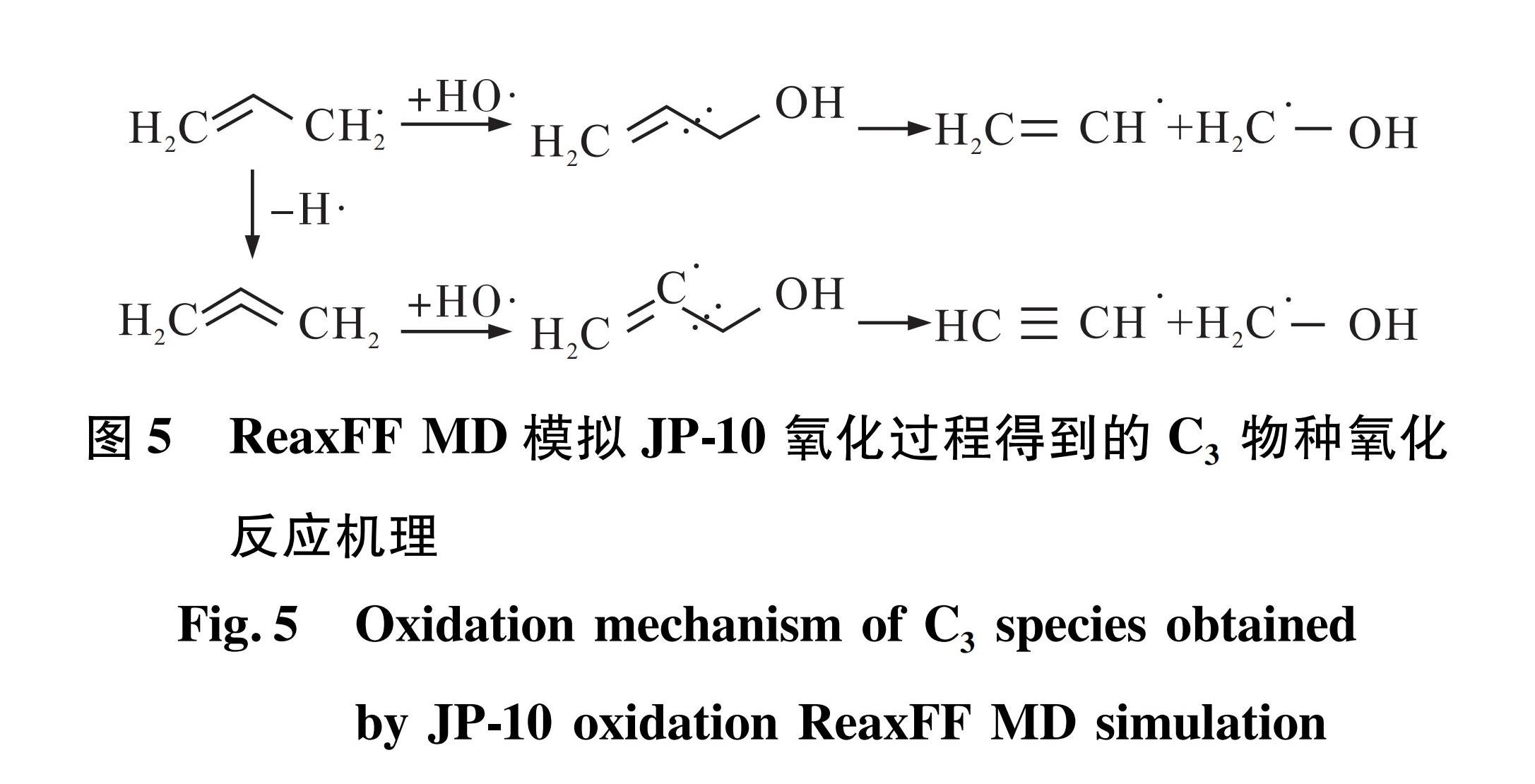
2)C3物种主要为C3H4(丙二烯)与C3H5(丙烯基),热解条件下C3H5容易发生脱氢反应生成C3H4或发生深度裂解反应生成CH3和C2H2。但在氧化条件下,模拟发现C3H5更易与HO反应生成相对稳定的产物C3H5OH(丙烯醇),如图5所示。C3H5OH在高温条件下会分解为C2H3和CH2OH(甲醇自由基),C2H3与CH2OH的氧化过程如图6与图7所示。
图5 ReaxFF MD模拟JP-10氧化过程得到的C3物种氧化反应机理
Fig.5 Oxidation mechanism of C3 species obtained by JP-10 oxidation ReaxFF MD simulation
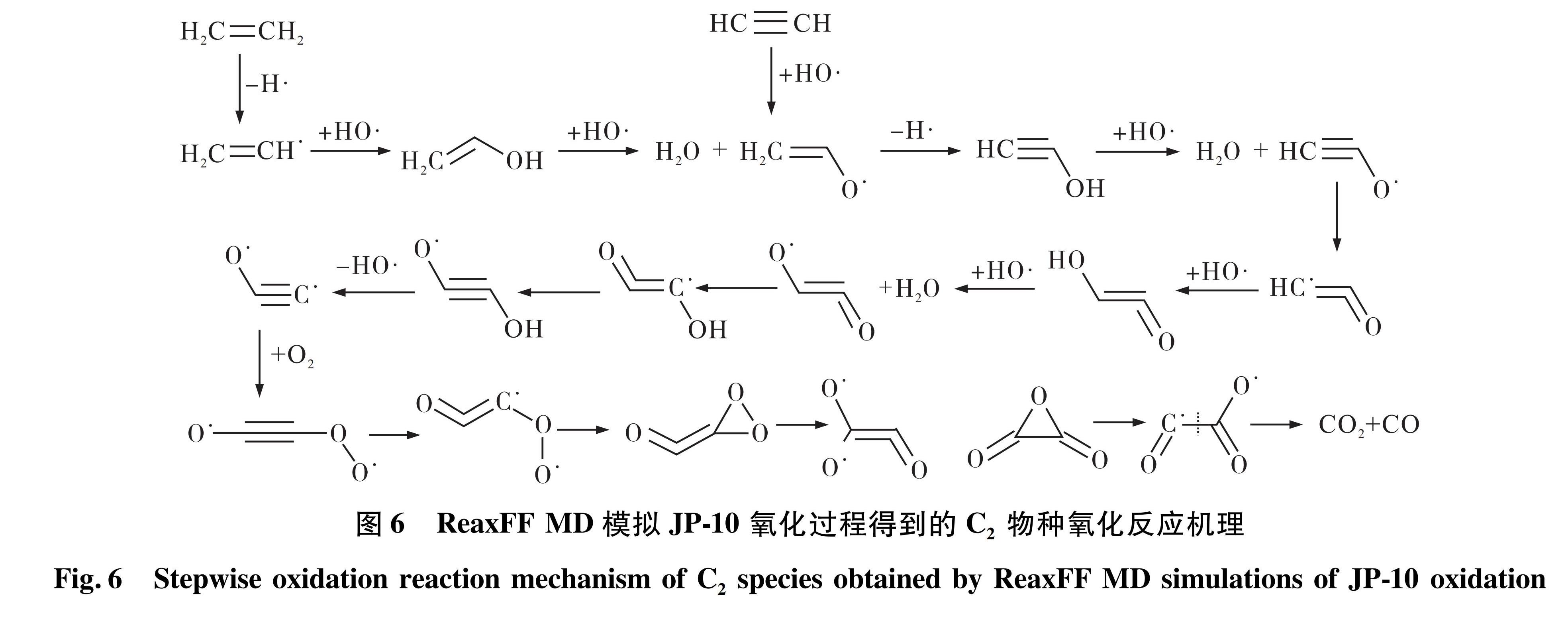
3)C2物种主要为C2H3(乙烯基)、C2H4(乙烯)和C2H2(乙炔),在高温热解条件下存在着C2H4脱氢生成C2H3、C2H3再脱氢生成C2H2的反应趋势; 而在氧化条件下,C2的氧化过程较为复杂,如图6所示。C2H3、C2H4和C2H2都会先生成CH2CHO(乙烯醇自由基),再经过脱氢反应生成CHCOH(乙炔醇),其与CH2CO(乙烯醛)互为异构体,能够相互转化。
图6 ReaxFF MD模拟JP-10氧化过程得到的C2物种氧化反应机理
Fig.6 Stepwise oxidation reaction mechanism of C2 species obtained by ReaxFF MD simulations of JP-10 oxidation
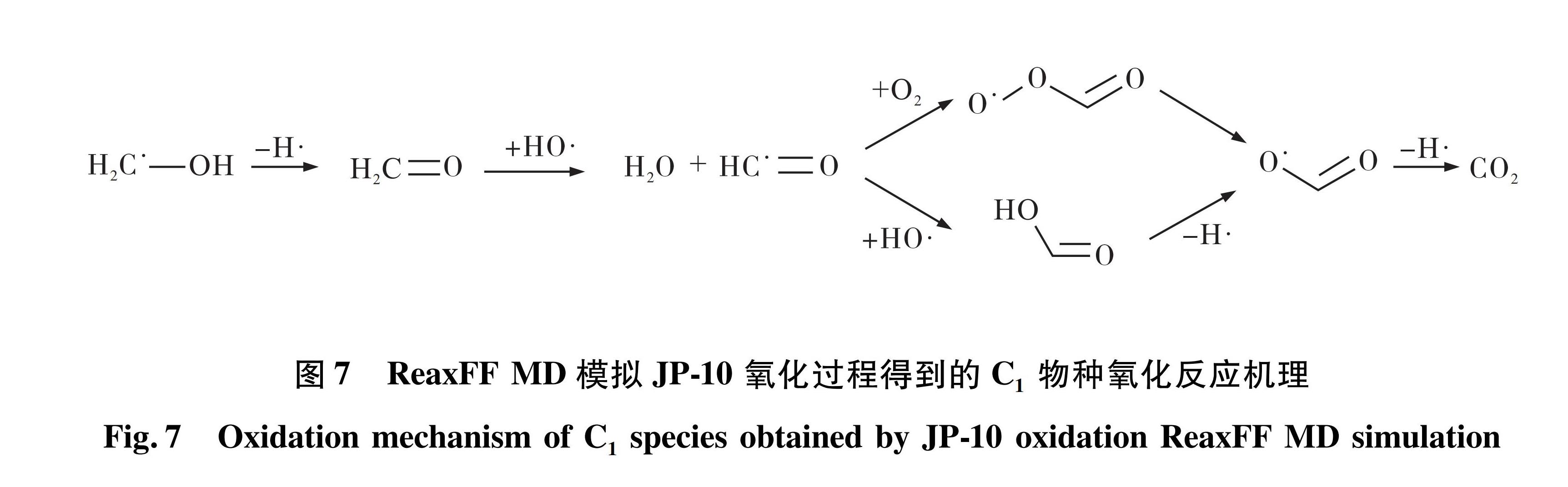
4)氧化模拟中C1物种主要为CH2OH,基本都由C3物种分解得到,其后续氧化路径如图7所示。与C2物种氧化类似,CH2OH依次发生了脱氢反应、HO加成反应、O2加成生成COO结构。不同的是,HO加成产物HCOOH(甲酸)发生脱氢反应以及O2加成产物HCOOO脱去O自由基均会生成HCOO(甲酸基),HCOO能直接脱氢生成CO2。
图7 ReaxFF MD模拟JP-10氧化过程得到的C1物种氧化反应机理
Fig.7 Oxidation mechanism of C1 species obtained by JP-10 oxidation ReaxFF MD simulation
从C0-C3物种的氧化机理得出JP-10的氧化反应过程可以概括为H与O2先生成HO2再生成HO,该过程是最主要的放热反应。H在整个氧化过程中循环生成与消耗,是链反应最初的引发剂,而HO是直接氧化C0-C3小分子烃的最主要的氧化剂; C3物种先与HO反应生成稳定产物C3H5OH(丙烯醇),再分解为C2与C1物种分别被氧化; C2分子的氧化路径是最复杂和重要的部分,经历了多步脱氢、HO加成、氢转移、O2加成、COO构型转变才生成H2O和CO2,其中CHCOH(乙炔醇)与异构体CH2CO(乙烯醛)为重要中间产物; C1分子氧化过程与C2类似,经过脱氢、HO加成、氢转移、O2加成反应后生成H2O和CO2。
2.4 模拟结果与文献对比
基于理论方法提出的最早的JP-10燃烧模型(San Diego燃烧模型)[26]包含36个物种和174个反应, 物种包括O2、H2、H2O、HO2、H2O2、H、OH、O、CO、CO2、CH4、CH3、CH3O、CH2OH、CH2O、CHO、singlet CH2、triplet CH2、CH、C2H、HCCO、CH2CO、CH2CHO、C2H2、C2H3、C2H4、C2H5、C2H6、C3H3、C3H4、C3H5、C3H6、C3H7、C4H6、C5H8和JP-10,其中大部分重要物种的类型与本工作模拟结果一致,但本文模拟得到的CH、CH2、C2H、C2H5、C2H6、C3H6和C3H7物种数量较少,原因是CH、CH2、C2H物种相对不稳定,且C2H5、C2H6、C3H6、C3H7物种H/C比较高。San Diego燃烧模型中C5以上的分子的裂解集总反应与C3及以下的烃类小分子的氧化反应与本文ReaxFF MD模拟结果一致。
Gao等[27]通过激波管实验与RMG反应路径生成结合的方法获得的JP-10燃烧反应机理模型约含700个物种、15 000个反应。JP-10分子热解阶段会生成包含环戊烯、环己烯和芳香环结构的分子,并伴随着C2H4、C2H3、C2H2、C3H5、H、H2分子或自由基的生成,其物种类型与本文的ReaxFF MD模拟结果基本一致。区别在于模拟得到的芳烃数量极少,含氧产物的反应路径有所不同,模拟发现JP-10还存在裂解生成C5、C8链烃分子的重要路径。
清华大学Zhong等[28]结合San Diego燃烧模型和Gao模型,通过燃烧弹实验获得的JP-10燃烧模型包括189个物种和1 287个反应。C5以上大分子的反应机理中包含C6H6分子脱氢、氧化生成C6H5O与CO的反应路径。其他热解阶段的产物主要为苯、环戊二烯、环戊烯、1,3-戊二烯、C4H6、C3H5、C2H4、C2H2、CH3和H,与本文结果一致。不同的是,文献[28]的敏感性分析结果显示层流火焰主要受C0-C3反应控制,CH3加H生成CH4的链终止反应较为重要,点火延迟则不仅由C0-C3控制,也与JP-10和热解初级产物的反应有关。与本文结果的差异主要体现在CH3的反应上,本文的JP-10裂解模拟发现CH3与H生成较晚,常见于JP-10已完全裂解成C5及以下大小的分子之后,相对于表4中的反应重要性较低。天津大学Jiang等[29]基于HyCHem理论[30]提出了改进后的JP-10的燃烧模型,包括127个物种和866个反应。本文模拟结果得到的JP-10热解主要产物中C6H6和C7H8分子数量较少,其他热解产物如H、CH3、C3H5、CH4、C2H4、C3H6、C5H6与文献[29]的模型基本一致。文献[29]认为其改进的模型预测能力体现为增加了醛类氧化中间产物(甲醛与乙醛),与本文ReaxFF MD结果中乙烯醛与甲醛分别是C2和C1氧化路径的重要物种大体一致。文献[29]通过层流火焰和点火延迟的敏感性分析指出H和O2生成HO以及CO和HO生成CO2和H的反应较为重要,本文模拟发现更多的机理细节,即JP-10氧化过程中H与O2先生成HO2、再生成OH,且生成HO2的反应释热量占主导地位,同时CO与HO生成CO2与H的反应也是重要的释热反应。
ReaxFF MD模拟获得的反应数量和种类繁多,尤其是在活性自由基较多的氧化体系中,因此模拟结果能较好地覆盖已有文献中提到过的物种与反应。但本文提出的反应机理是基于反应数量和能量排序所总结获得,并且为了减少计算代价设置的模拟温度和压力较高。因此本文仍存在与文献不一致的结果,如热解阶段芳香烃的生成反应、C5以上烃分子的氧化反应、CH3自由基的相关反应等。本工作的模拟结果在反应路径上也提供了新的认识,如C10物种到C5物种裂解的反应路径; C0机理中O2与H生成HO自由基的多步反应路径; C1-C3机理中C2机理相对复杂与重要。